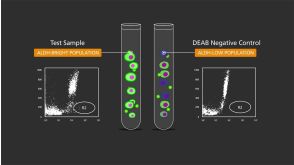
Effects of MLN518, a dual FLT3 and KIT inhibitor, on normal and malignant hematopoiesis.
Internal tandem duplications (ITDs) of the FMS-like tyrosine kinase 3 (FLT3) receptor tyrosine kinase are found in approximately 30% of patients with acute myelogenous leukemia (AML) and are associated with a poor prognosis. FLT3 ITD mutations result in constitutive kinase activation and are thought to be pathogenetically relevant,implicating FLT3 as a plausible therapeutic target. MLN518 (formerly CT53518) is a small molecule inhibitor of the FLT3,KIT,and platelet-derived growth-factor receptor (PDGFR) tyrosine kinases with significant activity in murine models of FLT3 ITD-positive leukemia. Given the importance of FLT3 and KIT for normal hematopoietic progenitor cells,we analyzed the effect of MLN518 on murine hematopoiesis under steady-state conditions,after chemotherapy-induced myelosuppression,and during bone marrow transplantation. In these assays,we show that MLN518 has mild toxicity toward normal hematopoiesis at concentrations that are effective in treating FLT3 ITD-positive leukemia in mice. We also demonstrate that MLN518 preferentially inhibits the growth of blast colonies from FLT3 ITD-positive compared with ITD-negative patients with AML,at concentrations that do not significantly affect colony formation by normal human progenitor cells. In analogy to imatinib mesylate in BCR-ABL-positive acute leukemia,MLN518-induced remissions may not be durable. Our studies provide the basis for integrating this compound into chemotherapy and transplantation protocols.
View Publication
产品类型:
产品号#:
03231
84434
84444
84534
84544
产品名:
MethoCult™M3231
文献
Hirano T et al. (DEC 2015)
Molecular Cancer 14 1 90
Long noncoding RNA, CCDC26, controls myeloid leukemia cell growth through regulation of KIT expression
BACKGROUND Accumulating evidence suggests that some long noncoding RNAs (lncRNAs) are involved in certain diseases,such as cancer. The lncRNA,CCDC26,is related to childhood acute myeloid leukemia (AML) because its copy number is altered in AML patients. RESULTS We found that CCDC26 transcripts were abundant in the nuclear fraction of K562 human myeloid leukemia cells. To examine the function of CCDC26,gene knockdown (KD) was performed using short hairpin RNAs (shRNAs),and four KD clones,in which CCDC26 expression was suppressed to 1% of its normal level,were isolated. This down-regulation included suppression of CCDC26 intron-containing transcripts (the CCDC26 precursor mRNA),indicating that transcriptional gene suppression (TGS),not post-transcriptional suppression,was occurring. The shRNA targeting one of the two CCDC26 splice variants also suppressed the other splice variant,which is further evidence for TGS. Growth rates of KD clones were reduced compared with non-KD control cells in media containing normal or high serum concentrations. In contrast,enhanced growth rates in media containing much lower serum concentrations and increased survival periods after serum withdrawal were observed for KD clones. DNA microarray and quantitative polymerase chain reaction screening for differentially expressed genes between KD clones and non-KD control cells revealed significant up-regulation of the tyrosine kinase receptor,KIT,hyperactive mutations of which are often found in AML. Treatment of KD clones with ISCK03,a KIT-specific inhibitor,eliminated the increased survival of KD clones in the absence of serum. CONCLUSIONS We suggest that CCDC26 controls growth of myeloid leukemia cells through regulation of KIT expression. A KIT inhibitor might be an effective treatment against the forms of AML in which CCDC26 is altered.
View Publication
产品类型:
产品号#:
03814
产品名:
ClonaCell™tcs介质
文献
Aichberger KJ et al. (DEC 2009)
Blood 114 26 5342--51
Identification of proapoptotic Bim as a tumor suppressor in neoplastic mast cells: role of KIT D816V and effects of various targeted drugs.
Systemic mastocytosis (SM) is a myeloid neoplasm involving mast cells (MCs) and their progenitors. In most cases,neoplastic cells display the D816V-mutated variant of KIT. KIT D816V exhibits constitutive tyrosine kinase (TK) activity and has been implicated in increased survival and growth of neoplastic MCs. Recent data suggest that the proapoptotic BH3-only death regulator Bim plays a role as a tumor suppressor in various myeloid neoplasms. We found that KIT D816V suppresses expression of Bim in Ba/F3 cells. The KIT D816-induced down-regulation of Bim was rescued by the KIT-targeting drug PKC412/midostaurin. Both PKC412 and the proteasome-inhibitor bortezomib were found to decrease growth and promote expression of Bim in MC leukemia cell lines HMC-1.1 (D816V negative) and HMC-1.2 (D816V positive). Both drugs were also found to counteract growth of primary neoplastic MCs. Furthermore,midostaurin was found to cooperate with bortezomib and with the BH3-mimetic obatoclax in producing growth inhibition in both HMC-1 subclones. Finally,a Bim-specific siRNA was found to rescue HMC-1 cells from PKC412-induced cell death. Our data show that KIT D816V suppresses expression of proapoptotic Bim in neoplastic MCs. Targeting of Bcl-2 family members by drugs promoting Bim (re)-expression,or by BH3-mimetics such as obatoclax,may be an attractive therapy concept in SM.
View Publication
Corbacioglu S et al. (NOV 2006)
Blood 108 10 3504--13
Newly identified c-KIT receptor tyrosine kinase ITD in childhood AML induces ligand-independent growth and is responsive to a synergistic effect of imatinib and rapamycin.
Activating mutations of c-KIT lead to ligand-independent growth. Internal tandem duplications (ITDs) of exon 11,which encodes the juxtamembrane domain (JMD),are constitutively activating mutations found in 7% of gastrointestinal stromal tumors (GISTs) but have not been described in childhood acute myeloid leukemia (AML). DNA and cDNA from 60 children with AML were screened by polymerase chain reaction (PCR) for mutations of the JMD. A complex ITD (kit cITD) involving exon 11 and exon 12 was identified with a relative frequency of 7% (4/60). The human kit cITDs were inserted into the murine c-Kit backbone and expressed in Ba/F3 cells. KIT cITD induced factorindependent growth and apoptosis resistance,and exhibited constitutive autophosphorylation. KIT cITD constitutively activated the PI3K/AKT pathway and phosphorylated STAT1,STAT3,STAT5,and SHP-2. Imatinib (IM) or rapamycin (Rap) led to complete inhibition of growth,with IC50 values at nanomolar levels. IM and Rap synergistically inhibited growth and surmounted KIT cITD-induced apoptosis resistance. IM but not LY294002 inhibited phosphorylation of STAT3 and STAT5,suggesting aberrant cross talk between PI3K- and STAT-activating pathways. The findings presented may have immediate therapeutic impact for a subgroup of childhood AML-expressing c-KIT mutations.
View Publication

 EasySep™小鼠TIL(CD45)正选试剂盒
EasySep™小鼠TIL(CD45)正选试剂盒




 BrochureElevate Tissue Dissociation with STEMprep™
BrochureElevate Tissue Dissociation with STEMprep™ 技术窍门In Vitro Drug Screening with the StemSpan™ Leukemic Cell Culture Kit
技术窍门In Vitro Drug Screening with the StemSpan™ Leukemic Cell Culture Kit 文献
文献 科学海报Optimized Capture and Purification of Nucleic Acid Using the EasySep™ Total Nucleic Acid Extraction Kit, A Robust Magnetic Particle-Based Extraction System
科学海报Optimized Capture and Purification of Nucleic Acid Using the EasySep™ Total Nucleic Acid Extraction Kit, A Robust Magnetic Particle-Based Extraction System