
 EasySep™小鼠TIL(CD45)正选试剂盒
EasySep™小鼠TIL(CD45)正选试剂盒




技术资料
-
 50:59
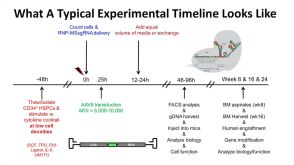
线上讲座Gene Targeting in Hematopoietic Stem Cells for Basic and Translational Research发布日期: 11/28/2018
50:59
线上讲座Gene Targeting in Hematopoietic Stem Cells for Basic and Translational Research发布日期: 11/28/2018 -
 31:39
线上讲座Serum- and Feeder-Free Differentiation of Erythroid Progenitor Cells from hPSCs发布日期: 05/21/2021
31:39
线上讲座Serum- and Feeder-Free Differentiation of Erythroid Progenitor Cells from hPSCs发布日期: 05/21/2021 -
 50:50
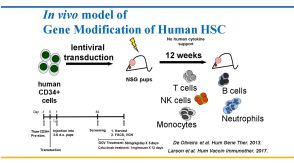
线上讲座Humanized Mouse Models for Hematopoietic Stem Cell Research: Principles and Pitfalls发布日期: 03/09/2018
50:50
线上讲座Humanized Mouse Models for Hematopoietic Stem Cell Research: Principles and Pitfalls发布日期: 03/09/2018





 沪公网安备31010102008431号
沪公网安备31010102008431号