Yamashita J et al. (NOV 2000)
Nature 408 6808 92--6
Flk1-positive cells derived from embryonic stem cells serve as vascular progenitors.
Interaction between endothelial cells and mural cells (pericytes and vascular smooth muscle) is essential for vascular development and maintenance. Endothelial cells arise from Flk1-expressing (Flk1+) mesoderm cells,whereas mural cells are believed to derive from mesoderm,neural crest or epicardial cells and migrate to form the vessel wall. Difficulty in preparing pure populations of these lineages has hampered dissection of the mechanisms underlying vascular formation. Here we show that Flk1+ cells derived from embryonic stem cells can differentiate into both endothelial and mural cells and can reproduce the vascular organization process. Vascular endothelial growth factor promotes endothelial cell differentiation,whereas mural cells are induced by platelet-derived growth factor-BB. Vascular cells derived from Flk1+ cells can organize into vessel-like structures consisting of endothelial tubes supported by mural cells in three-dimensional culture. Injection of Flk1+ cells into chick embryos showed that they can incorporate as endothelial and mural cells and contribute to the developing vasculature in vivo. Our findings indicate that Flk1+ cells can act as 'vascular progenitor cells' to form mature vessels and thus offer potential for tissue engineering of the vascular system.
View Publication
产品号#:
06902
06952
00321
00322
00323
00324
00325
产品名:
Ban K et al. (OCT 2013)
Circulation 128 17 1897--1909
Purification of cardiomyocytes from differentiating pluripotent stem cells using molecular beacons that target cardiomyocyte-specific mRNA
BACKGROUND: Although methods for generating cardiomyocytes from pluripotent stem cells have been reported,current methods produce heterogeneous mixtures of cardiomyocytes and noncardiomyocyte cells. Here,we report an entirely novel system in which pluripotent stem cell-derived cardiomyocytes are purified by cardiomyocyte-specific molecular beacons (MBs). MBs are nanoscale probes that emit a fluorescence signal when hybridized to target mRNAs.backslashnbackslashnMETHOD AND RESULTS: Five MBs targeting mRNAs of either cardiac troponin T or myosin heavy chain 6/7 were generated. Among 5 MBs,an MB that targeted myosin heavy chain 6/7 mRNA (MHC1-MB) identified up to 99% of HL-1 cardiomyocytes,a mouse cardiomyocyte cell line,but textless3% of 4 noncardiomyocyte cell types in flow cytometry analysis,which indicates that MHC1-MB is specific for identifying cardiomyocytes. We delivered MHC1-MB into cardiomyogenically differentiated pluripotent stem cells through nucleofection. The detection rate of cardiomyocytes was similar to the percentages of cardiac troponin T- or cardiac troponin I-positive cardiomyocytes,which supports the specificity of MBs. Finally,MHC1-MB-positive cells were sorted by fluorescence-activated cell sorter from mouse and human pluripotent stem cell differentiating cultures,and ≈97% cells expressed cardiac troponin T or cardiac troponin I as determined by flow cytometry. These MB-based sorted cells maintained their cardiomyocyte characteristics,which was verified by spontaneous beating,electrophysiological studies,and expression of cardiac proteins. When transplanted in a myocardial infarction model,MB-based purified cardiomyocytes improved cardiac function and demonstrated significant engraftment for 4 weeks without forming tumors.backslashnbackslashnCONCLUSIONS: We developed a novel cardiomyocyte selection system that allows production of highly purified cardiomyocytes. These purified cardiomyocytes and this system can be valuable for cell therapy and drug discovery.
View Publication
产品号#:
05850
05857
05870
05875
85850
85857
85870
85875
产品名:
mTeSR™1
mTeSR™1
Huber BC et al. (NOV 2013)
STEM CELLS 31 11 2354--2363
Costimulation-adhesion blockade is superior to Cyclosporine A and prednisone immunosuppressive therapy for preventing rejection of differentiated human embryonic stem cells following transplantation
RATIONALE: Human embryonic stem cell (hESC) derivatives are attractive candidates for therapeutic use. The engraftment and survival of hESC derivatives as xenografts or allografts require effective immunosuppression to prevent immune cell infiltration and graft destruction.backslashnbackslashnOBJECTIVE: To test the hypothesis that a short-course,dual-agent regimen of two costimulation-adhesion blockade agents can induce better engraftment of hESC derivatives compared to current immunosuppressive agents.backslashnbackslashnMETHODS AND RESULTS: We transduced hESCs with a double fusion reporter gene construct expressing firefly luciferase (Fluc) and enhanced green fluorescent protein,and differentiated these cells to endothelial cells (hESC-ECs). Reporter gene expression enabled longitudinal assessment of cell engraftment by bioluminescence imaging. Costimulation-adhesion therapy resulted in superior hESC-EC and mouse EC engraftment compared to cyclosporine therapy in a hind limb model. Costimulation-adhesion therapy also promoted robust hESC-EC and hESC-derived cardiomyocyte survival in an ischemic myocardial injury model. Improved hESC-EC engraftment had a cardioprotective effect after myocardial injury,as assessed by magnetic resonance imaging. Mechanistically,costimulation-adhesion therapy is associated with systemic and intragraft upregulation of T-cell immunoglobulin and mucin domain 3 (TIM3) and a reduced proinflammatory cytokine profile.backslashnbackslashnCONCLUSIONS: Costimulation-adhesion therapy is a superior alternative to current clinical immunosuppressive strategies for preventing the post-transplant rejection of hESC derivatives. By extending the window for cellular engraftment,costimulation-adhesion therapy enhances functional preservation following ischemic injury. This regimen may function through a TIM3-dependent mechanism.
View Publication
产品号#:
05850
05857
05870
05875
85850
85857
85870
85875
产品名:
mTeSR™1
mTeSR™1
Cheng Y et al. ( 2013)
BMC cell biology 14 1 44
Physiological β-catenin signaling controls self-renewal networks and generation of stem-like cells from nasopharyngeal carcinoma.
BACKGROUND: A few reports suggested that low levels of Wnt signaling might drive cell reprogramming,but these studies could not establish a clear relationship between Wnt signaling and self-renewal networks. There are ongoing debates as to whether and how the Wnt/β-catenin signaling is involved in the control of pluripotency gene networks. Additionally,whether physiological β-catenin signaling generates stem-like cells through interactions with other pathways is as yet unclear. The nasopharyngeal carcinoma HONE1 cells have low expression of β-catenin and wild-type expression of p53,which provided a possibility to study regulatory mechanism of stemness networks induced by physiological levels of Wnt signaling in these cells.backslashnbackslashnRESULTS: Introduction of increased β-catenin signaling,haploid expression of β-catenin under control by its natural regulators in transferred chromosome 3,resulted in activation of Wnt/β-catenin networks and dedifferentiation in HONE1 hybrid cell lines,but not in esophageal carcinoma SLMT1 hybrid cells that had high levels of endogenous β-catenin expression. HONE1 hybrid cells displayed stem cell-like properties,including enhancement of CD24(+) and CD44(+) populations and generation of spheres that were not observed in parental HONE1 cells. Signaling cascades were detected in HONE1 hybrid cells,including activation of p53- and RB1-mediated tumor suppressor pathways,up-regulation of Nanog-,Oct4-,Sox2-,and Klf4-mediated pluripotency networks,and altered E-cadherin expression in both in vitro and in vivo assays. qPCR array analyses further revealed interactions of physiological Wnt/β-catenin signaling with other pathways such as epithelial-mesenchymal transition,TGF-β,Activin,BMPR,FGFR2,and LIFR- and IL6ST-mediated cell self-renewal networks. Using β-catenin shRNA inhibitory assays,a dominant role for β-catenin in these cellular network activities was observed. The expression of cell surface markers such as CD9,CD24,CD44,CD90,and CD133 in generated spheres was progressively up-regulated compared to HONE1 hybrid cells. Thirty-four up-regulated components of the Wnt pathway were identified in these spheres.backslashnbackslashnCONCLUSIONS: Wnt/β-catenin signaling regulates self-renewal networks and plays a central role in the control of pluripotency genes,tumor suppressive pathways and expression of cancer stem cell markers. This current study provides a novel platform to investigate the interaction of physiological Wnt/β-catenin signaling with stemness transition networks.
View Publication
Biophysical regulation of epigenetic state and cell reprogramming
Biochemical factors can help reprogram somatic cells into pluripotent stem cells,yet the role of biophysical factors during reprogramming is unknown. Here,we show that biophysical cues,in the form of parallel microgrooves on the surface of cell-adhesive substrates,can replace the effects of small-molecule epigenetic modifiers and significantly improve reprogramming efficiency. The mechanism relies on the mechanomodulation of the cells' epigenetic state. Specifically,decreased histone deacetylase activity and upregulation of the expression of WD repeat domain 5 (WDR5)—a subunit of H3 methyltranferase—by microgrooved surfaces lead to increased histone H3 acetylation and methylation. We also show that microtopography promotes a mesenchymal-to-epithelial transition in adult fibroblasts. Nanofibrous scaffolds with aligned fibre orientation produce effects similar to those produced by microgrooves,suggesting that changes in cell morphology may be responsible for modulation of the epigenetic state. These findings have important implications in cell biology and in the optimization of biomaterials for cell-engineering applications.
View Publication
产品号#:
05850
05857
05870
05875
85850
85857
85870
85875
产品名:
mTeSR™1
mTeSR™1
Chen A et al. (JAN 2014)
Biomaterials 35 2 675--683
Integrated platform for functional monitoring of biomimetic heart sheets derived from human pluripotent stem cells
We present an integrated platform comprised of a biomimetic substrate and physiologically aligned human pluripotent stem cell-derived cardiomyocytes (CMs) with optical detection and algorithms to monitor subtle changes in cardiac properties under various conditions. In the native heart,anisotropic tissue structures facilitate important concerted mechanical contraction and electrical propagation. To recapitulate the architecture necessary for a physiologically accurate heart response,we have developed a simple way to create large areas of aligned CMs with improved functional properties using shrink-wrap film. Combined with simple bright field imaging,obviating the need for fluorescent labels or beads,we quantify and analyze key cardiac contractile parameters. To evaluate the performance capabilities of this platform,the effects of two drugs,E-4031 and isoprenaline,were examined. Cardiac cells supplemented with E-4031 exhibited an increase in contractile duration exclusively due to prolonged relaxation peak. Notably,cells aligned on the biomimetic platform responded detectably down to a dosage of 3nm E-4031,which is lower than the IC50 in the hERG channel assay. Cells supplemented with isoprenaline exhibited increased contractile frequency and acceleration. Interestingly,cells grown on the biomimetic substrate were more responsive to isoprenaline than those grown on the two control surfaces,suggesting topography may help induce more mature ion channel development. This simple and low-cost platform could thus be a powerful tool for longitudinal assays as well as an effective tool for drug screening and basic cardiac research. ?? 2013 Elsevier Ltd.
View Publication

 EasySep™小鼠TIL(CD45)正选试剂盒
EasySep™小鼠TIL(CD45)正选试剂盒




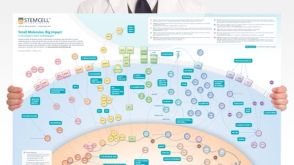 挂图Small Molecules, Big Impact in Pluripotent Stem Cell Research Overview of signaling pathways and small molecules in pluripotent stem cell research
挂图Small Molecules, Big Impact in Pluripotent Stem Cell Research Overview of signaling pathways and small molecules in pluripotent stem cell research 挂图Stem Cell States: Naive to Primed Pluripotency Properties of naive (ground) and primed pluripotent stem cells
挂图Stem Cell States: Naive to Primed Pluripotency Properties of naive (ground) and primed pluripotent stem cells


 科学海报STEMdiff™ Cerebral Organoid Kit: A New Tool for the Culture of 3D Brain Organoids Derived from hPSCs
科学海报STEMdiff™ Cerebral Organoid Kit: A New Tool for the Culture of 3D Brain Organoids Derived from hPSCs
 沪公网安备31010102008431号
沪公网安备31010102008431号