
 EasySep™小鼠TIL(CD45)正选试剂盒
EasySep™小鼠TIL(CD45)正选试剂盒




人原代T细胞的基因组编辑
人原代T细胞的基因组编辑
- Document # 27155
- Version 2.0.2
- 6/1/23
引言
CRISPR-Cas9是一种RNA引导的基因组编辑技术,能够轻松有效地对哺
乳动物细胞进行遗传操作,因此它对细胞生物学的研究具有划时代的意
义。通过对特定基因或调节区域的靶向修饰,研究人员现在可以快速生
成精确的遗传模型,以研究正常和患病的细胞生理学。除了用于研究目的
的基因操作之外,CRISPR-Cas9基因组编辑技术还在临床治疗领域有巨大
的潜力,包括免疫治疗和再生医学。以下内容阐述了针对人原代T细胞的
CRISPR-Cas9基因组编辑的说明,包括编辑前和编辑后培养条件的优化以
及评估基因组编辑效率的方法。
图1展示了CRISPR-Cas9基因组编辑的机制。简单来说,通过相关的CRISPR
RNA(crRNA)在与原间隔序列临近基序(PAM)(化脓性链球菌Cas9为
NGG)上游5’位点的碱基互补配对,将Cas9内切核酸酶靶向目的基因组
位点。crRNA与反式激活CRISPR RNA(tracrRNA)结合为与Cas9相互作用
的二聚体,以形成向导RNA(gRNA)。Cas9与gRNA形成复合物后,即可
靶向目的基因组位点,并在PAM序列的上游进行切割,产生3至4个碱基
的DNA断裂。接着内源性DNA修复系统开始修复断裂的DNA。非同源末
端连接(NHEJ)介导了断裂DNA分子的直接连接,由于NHEJ的易出错特
性,这可导致插入或缺失(INDEL)突变的形成。如果提供供体DNA序列
模板,则可以通过同源定向修复(HDR)途径实现精确DNA序列的敲入(
图1)。
尽管这项技术已成功应用于很多细胞系中,但在原代人免疫细胞中的应
用却遇到了CRISPR-Cas9组分的高效传递和表达这两项极大的挑战。早
期尝试将CRISPR-Cas9用于原代人T细胞的基因组编辑时,使用了病毒载
体1,2
或质粒3,4表达Cas9和gRNA,导致低靶向效率和高毒性。由于不想要
的基因突变和免疫原性的风险,这些表达系统也可能影响临床转化的安
全性。最近,用重组蛋白Cas9和体外转录(IVT)或合成gRNA制备的核糖
核蛋白(RNP)复合物对活化的T细胞进行电转,已在许多靶标中实现了高
功效6-10。在许多细胞类型中,包括人T细胞,体外转录的gRNA都会引起高
细胞毒性,这是因为5’三磷酸单链RNA(ssRNA)激活了I型干扰素介导的
免疫反应11。因此,基于RNP的方法会优先选择使用合成gRNA。ArciTect™
是基于RNP的CRISPR-Cas9表达系统,包括定制的合成gRNA,旨在完全支
持人原代T细胞的基因组编辑。
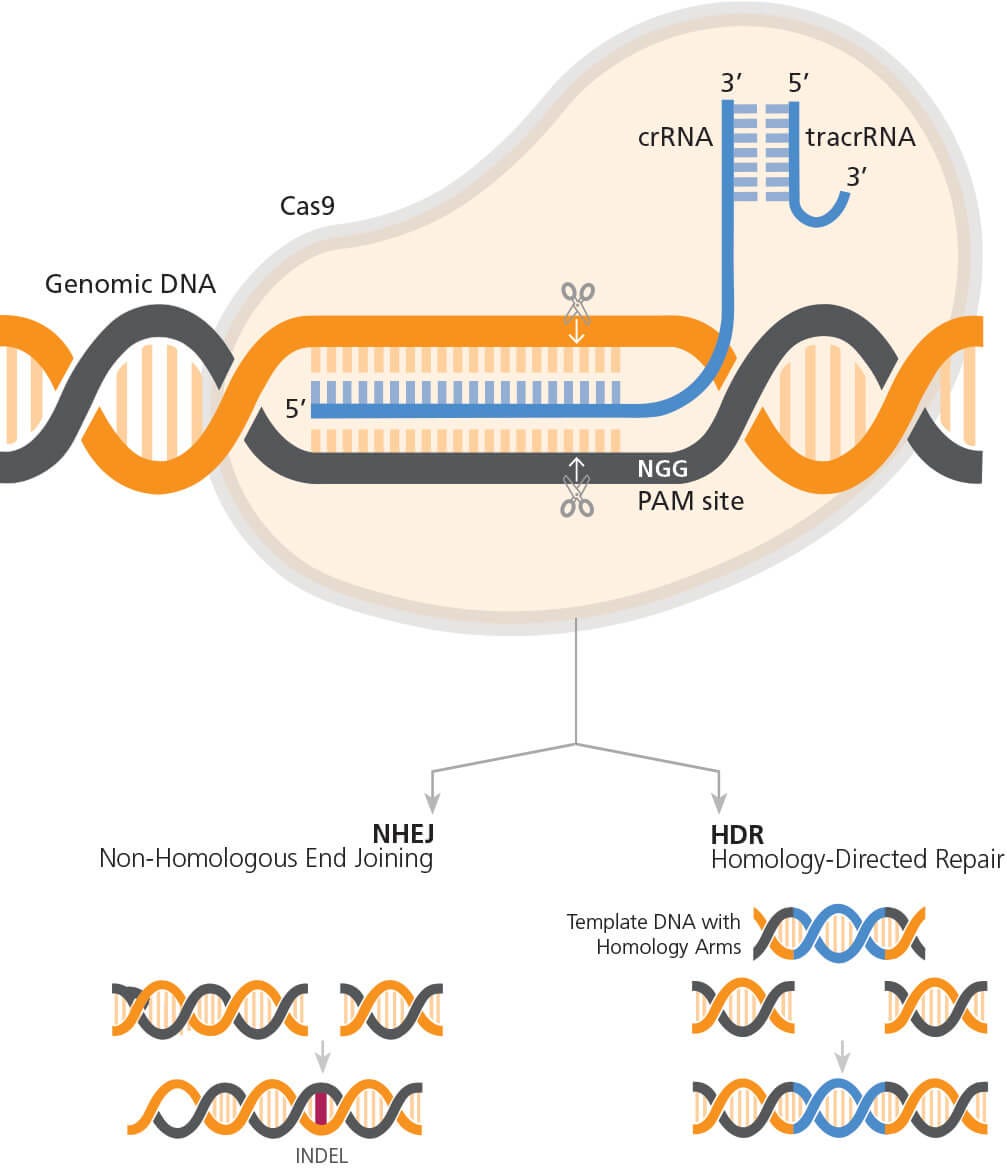
图1. CRISPR-Cas9 基因组编辑的机制
RNP复合物(通过Cas9和gRNA的合成)定向相对于靶点和PAM位点。Cas9在PAM序列 上游产生3-4 bp的DNA断裂,触发内源性DNA修复途径。如果通过NHEJ途径进行修复 则可能导致INDEL的形成,或者如果在实验设计中包含模板DNA,则可能通过HDR途径 导致精准序列的敲入。
除CRISPR-Cas9表达方法外,用于扩增和激活人原代T细胞的培养系统
也是成功进行基因组编辑的关键要素,因为在大多数实验中都需要激
活细胞12。尽管可以使用多种分离技术从不同样本来源分离T细胞,但
迄今为止,涉及T细胞的大多数基因组编辑研究都使用免疫磁珠分选
技术从外周血单个核细胞(PBMC)中分离的人原代T细胞7, 12-14
通过对表达和传递方法的改进,CRISPR-Cas9基因组编辑现已被迅速
纳入下一代免疫疗法的开发中。比如:嵌合抗原受体(CAR)T细胞疗
法,其中CRISPR-Cas9能够产生与适用于患者输入的同种异体免疫效
应细胞15。因此,可以通过敲除内源性T细胞受体(TCR)和人白细胞
抗原(HLA)I类分子来改造T细胞,以避免免疫排斥反应,在抑制性受
体(如程序性死亡1(PD-1)或细胞毒性T淋巴细胞抗原蛋白4(CTLA-1
))缺失的情况下,这种经改造后的T细胞效率会更高9-10,16-17。接着将
CAR构建体靶向内源TCR α恒定区(TRAC)基因位点18,以产生通用的
同种异体CAR-T细胞。
以下操作流程阐述了有关人原代T细胞的分离与激活,CRISPR-Cas9
RNP复合物的制备以及使用电转将RNP复合物导入激活的原代T细胞
中的步骤(图2)。这里我们将介绍一种生成基因敲除原代T细胞的高
效方法,以及有关在多个激活条件下对TCRαβ基因座进行CRISPRCas9基因组编辑的数据。查看本文档的讨论部分,了解替代策略、预
期结果以及对流程的修改。
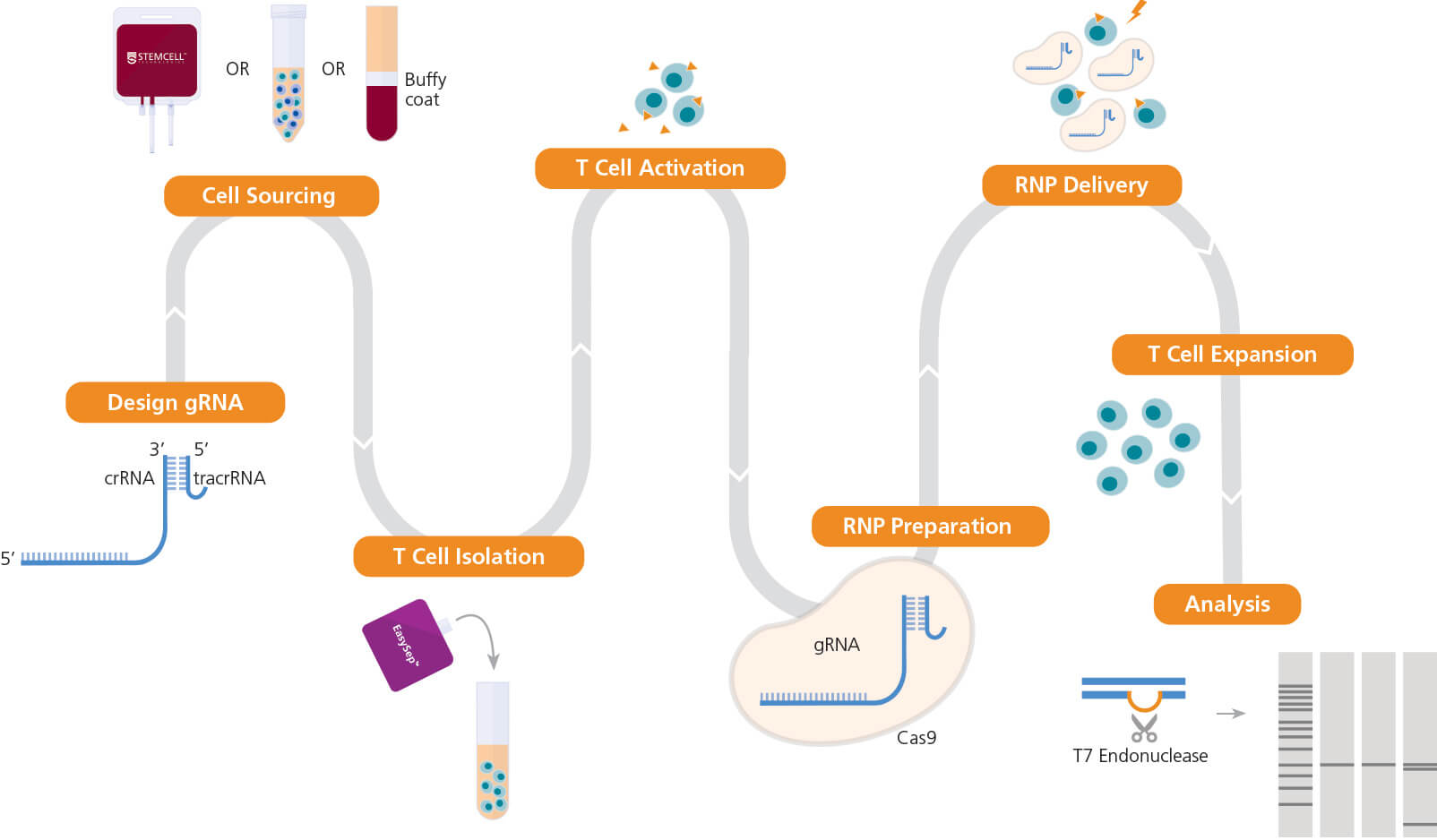
图2. T细胞基因组编辑的实验流程
一旦确定了用于编辑的目标基因座,开始设计向导RNA(gRNA)序列。人原代T细胞可以直接从STEMCELL购买,或通过使用STEMCELL无柱细胞分选技术(包括EasySep™)从包 括全血、白膜层、白细胞单采术样本或外周血单个核细胞(PBMC)等多种来源分离获得。接着,使用ImmunoCult™ CD3/CD28 T细胞激活剂或ImmunoCult™ CD3/CD28/CD2 T 细胞激活剂来活化T细胞。然后准备ArciTect™ CRISPR-Cas9核糖核蛋白(RNP)复合物,通过电转的方式将RNP复合物导入T细胞中,接下来使用ImmunoCult™-XF T细胞扩增培 养基扩增细胞。如果实验设计允许,可以使用ArciTect™ T7核酸内切酶I试剂盒(产品号#76021)或使用流式的方法检测编辑效率。橙色三角形表示ImmunoCult™人T细胞激活 剂。
在原代人T细胞上进行CRISPR-Cas9基因组编辑
所需材料
- ArciTect™ tracrRNA
- ArciTect™ 退火缓冲液(5X)
- 重悬缓冲液T
- 电解缓冲液E2
A. T细胞分选和激活
- 使用EasySep™人T细胞分选试剂盒(产品号 #70500)从外周血分
离人T细胞。具体操作请参考产品说明书(PIS,#DX20004)
可选:此外,可使用冷冻原代T细胞(产品号 #70024)。具体信息 请参考产品说明书(PIS,#DX27569)。 - 对细胞进行计数并将其浓度调整为1 x 106cells/mL后,接种至含 有2 mM L-谷氨酰胺,50 μg/mL庆大霉素和10 ng/mL(特定批次 为130 IU/mL)人重组IL-2细胞因子的ImmunoCult™-XF T细胞扩 增培养基中。
- 加入25 μL/mL的ImmunoCult™人CD3/CD28 T细胞激活剂来激活 T细胞。在37℃和5% CO2条件下孵育细胞悬液72小时。
- 可选:通过将激活标志物CD25与抗人CD25抗体(克隆号 BC96,产品号 #60158)结合并进行流式细胞术来测定T细胞的 激活状态。
B. 准备ArciTect™ crRNA和ArciTect™ tracrRNA的母液
- 打开ArciTect™ crRNA和ArciTect™ tracrRNA前简单离心。
- 按照表1,加入无核酸酶水制备终浓度为200μM的试剂。
- 混合均匀。如果不立即使用,可进行分装并在-80℃下储存长达1 个月。解冻后,应立即使用。切忌反复冻融。
表1. 制备200μM* ArciTect™ crRNA 或 ArciTect™ tracrRNA
*200 μM等同于200 pmol/μL
准备ArciTect™ CRISPR-Cas9 RNP复合物
- 如表2所示,在RNase-free微量离心管中混合各个组分以制备 60μM gRNA。该体积足以进行一次电转实验,请根据需要调整 试剂用量。混合均匀。
- 在热循环仪或加热块中,将gRNA混合物在95℃下孵育5分钟,然
后在60℃下孵育1分钟。冷却至室温(15-25℃),再置于冰上。
注意:如果不立即使用,可在-80℃下保存长达1个月。 - 如表3所示,在RNase-free微量离心管中混合60 μM gRNA(步骤 2中制备)和ArciTect™ Cas9核酸酶(或ArciTect™ Cas9-eGFP) 以制备RNP复合物。这些试剂足够进行一个孔的电转,如果需要 进行更多电转,请按比例调整体积。
- 轻轻上下吹打两次以混合RNP复合物混合物,避免试管内产生 气泡。
- 室温(15 - 25℃)孵育RNP复合物混合物10-15分钟。
表2. 制备60μM gRNA
表3. 制备RNP复合物混合物
如果使用3μg/uL ArciTect™ Cas9-eGFP核酸酶,对于每次实验所需的20μL体积,加 入11.4μL Cas9-eGFP到6 μL的60 μM gRNA和2.6μL重悬缓冲液T中。
D. 使用RNP复合物对T细胞进行电转
- 对一次电转(包括阳性和阴性对照),准备含有2 mM L-谷氨酰 胺,50μg/mL庆大霉素和10 ng/mL人重组IL-2细胞因子的2 mL的 ImmunoCult™-XF T细胞扩增培养基。
- 对每一次电转,在6孔板的每孔中加入2 mL于步骤1中准备的补 充培养基,并在37℃下进行孵育。剩余的补充培养基置于2 - 8℃ 下保存。
- 在15 mL锥形管中加入1.2 x 106被激活的T细胞悬液(经步骤A制 备),于室温下以300 x g离心5分钟。
- 吸除培养基并在100μL重悬液T中重悬细胞。
- 对每一次电转使用单独的RNase-free离心管,按照表4将RNP复 合物混合物(经步骤C制备)与激活的T细胞混合。
- 用移液枪轻轻上下吹打两次,避免试管内产生气泡。
注意:如果有气泡,电转时细胞的活力和转染效率会显著下降。
注意:参考电转仪器生产商的说明书。针对不同的细胞系可能需 要进行电转条件的优化。 - 使用100μL Neon®枪头,吸取100μL混合物,放入具有3mL电解 缓冲液E2的电转室中。使用表5中的设置对混合物进行电转。
- 电转结束后,立即将电转的细胞加入步骤2准备的6孔板的一个 孔中,于37℃和5% CO2下孵育2-3小时。
- 对细胞进行计数并将细胞浓度调整为2.5 x 105活细胞/mL,接种 至含有人重组IL-2细胞因子的ImmunoCult™-XF T细胞扩增培养 基中(步骤1中制备)。
- 可选:若使用Cas9-eGFP核酸酶,可以通过流式在电转后12 - 24 小时评估转染效率。
- 电转后,细胞需在37℃和5% CO2下孵育48 - 72小时,以便基因 编辑发生。收集细胞,评估基因组编辑效率。可使用ArciTect™高 保真DNA聚合酶试剂盒(产品号 #76026)和针对靶点区域附近 的引物对基因组DNA进行PCR扩增,然后对PCR扩增后的产物进 行测序。或者,可以使用ArciTect™ T7核酸内切酶I试剂盒(产品 号 #76021)对经PCR扩增后的产物进行编辑效率(% INDEL形成 率)的评估。有关详细信息,请参阅技术手册:评估基因组编辑效 率(文档号 #27126),可从我们的网站 www.stemcell.com获 取或联系我们索取副本。
表4. RNP混合物和T细胞混合用于电转
表5. 使用Neon®对人T细胞进行电转的推荐条件
案例分析:评估对人原代T细胞高效敲除TRAC的最佳培养方法
为了证明使用ArciTect™ CRISPR-Cas9系统的基因敲除,我们使用上文中描述的方法靶向TRAC位点。此外,我们测试了多种T细胞激活剂及其动力学
以找出具有最高敲除效率的体系。通过细胞表面没有TCR表达可轻松识别该位点由于INDEL引起的功能缺失。最重要的是通过靶向此基因座产生用
于CAR T细胞疗法的通用型同种异体T细胞18。
首先,我们设计了多个crRNAs(图3A,D),因为不同的crRNA序列可以在靶位点上表现出不同的效率(请参阅技术公告:设计Cas9 RNP直接传递的
基因组编辑的注意事项,文档号 #27083)。我们测试了4种crRNA在激活的人T细胞中的效率,并且通过流式细胞仪检测了TRAC敲除效率,以及通
过ArciTect™ T7核酸内切酶I试剂盒检测了INDEL的生成。所有的这四个crRNAs均显着降低了TCRαβ+细胞信号的频率(图3B),并在T7核酸内切酶I
分析(图3C)中显示出切割条带,从而证实了存在由于INDELs而导致的基因组错配。根据以上结果,crRNA2在流式细胞分析中显示出最高的敲除效
率,因此其被用于进一步的研究
接着,我们使用已经验证的crRNA2来比较各种T细胞激活条件下的敲
除效率。首先,从四个独立捐献者的外周血中分离出T细胞。分离出的
细胞在含有ImmunoCult™人CD3/CD28或CD3/CD28/CD2 T细胞激活
剂的Immunocult™-XF T细胞扩增培养基中培养2或3天,总共四个测
试条件。在第0天,大约10-20%的细胞表达了激活标记物CD25(数据
未显示),在激活2或3天后,其表达增加到80%以上(图4)。相比于
CD3/CD28 T细胞激活剂,CD3/CD28/CD2 T细胞激活剂的激活效果更
好(图4)。细胞被激活3天后,活化细胞的百分比更大(图4)。欲了解
更多关于T细胞激活和扩增条件优化的详情,请参考技术公告:人T细
胞扩增流程优化:早期细胞稀释的影响(文档号 #27143),可从我们
的网站www.stemcell.com获取或联系我们索取副本。
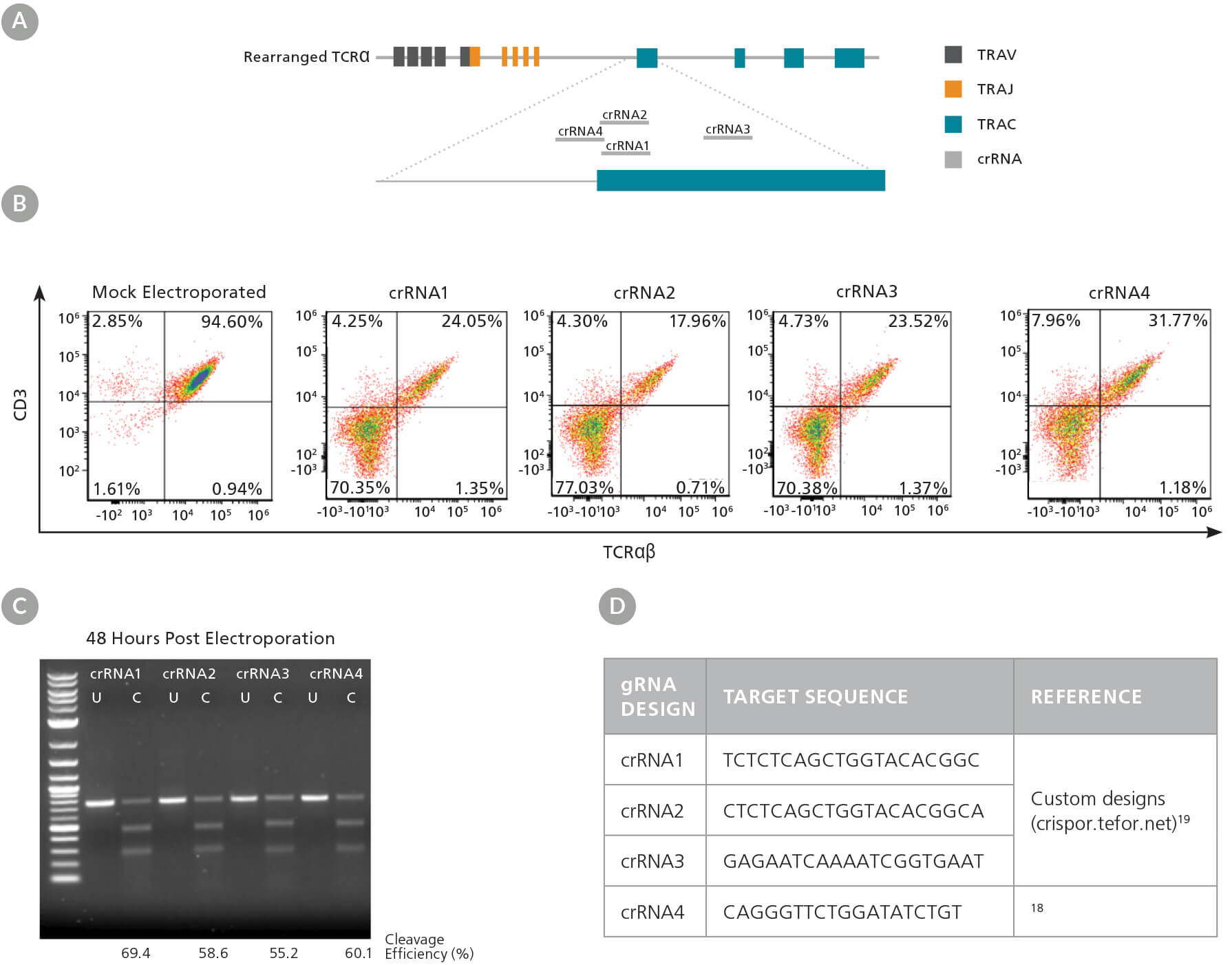
图3. TRAC gRNA的设计与评估
(A)crRNA序列1 - 4与TRAC第一个外显子对齐的TRAC基因位点示意图。(B)在RNP复合物电转(含相应的crRNA1-4)入激活的T细胞后48小时的TCRαβ/CD3流式结果。(C)使 用ArciTect™ T7核酸内切酶I试剂盒在电转后48小时评估基因组编辑(切割)的效率。U: 未切割; C: 已切割。(D)用于TRAC敲除的gRNA靶序列。
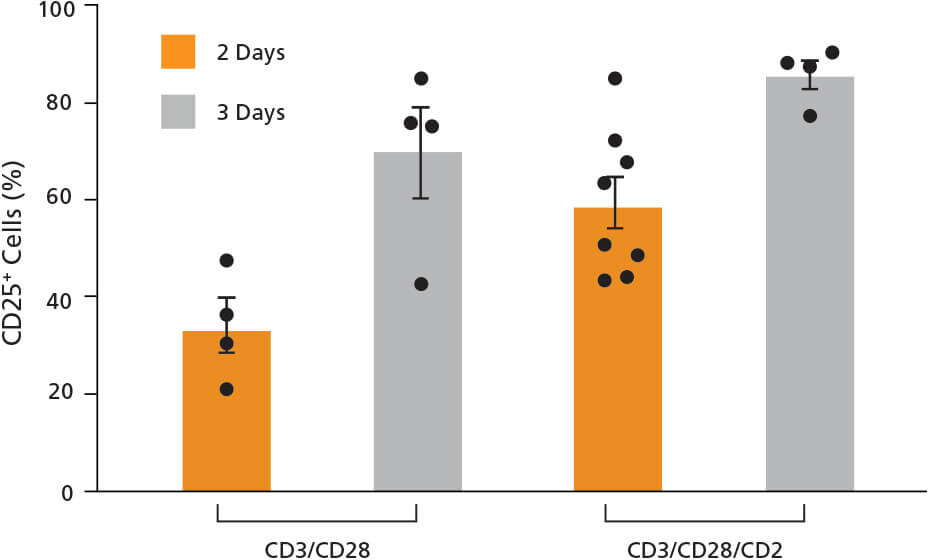
图4. T细胞经ImmunoCult™人T细胞激活剂处理后,表达CD25激活标 记物。
使用ImmunoCult™人CD3/CD28或CD3/CD28/CD2 T细胞激活剂活化分离的人T细 胞2或3天。通过CD25测定其活化状态。每个条件的每个数据点代表一个单独的供 体;n=4 - 8个供体。每一列误差线表示平均值 ± 标准差。
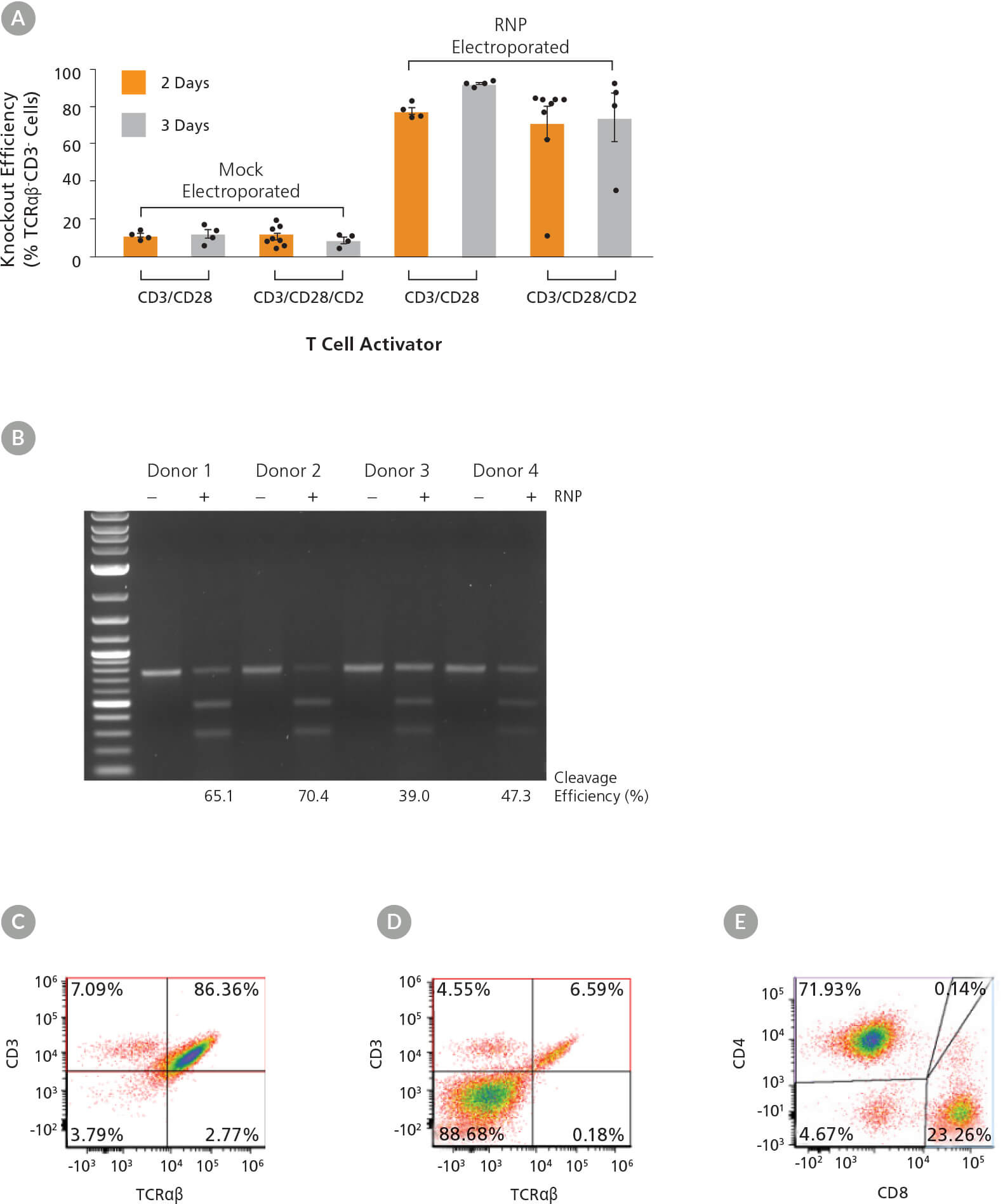
图5. 不同激活条件和动力学下对TRAC的高效敲除
(A)使用ImmunoCult™人CD3/CD28或CD3/CD28/CD2 T细胞激活剂活化人T细胞2 - 3天后,通过将TCRαβ和CD3受体与抗体结合,进行流式分析,来测定TRAC的敲除效率。每 个条件的每个数据点代表一个单独的供体;n=4 - 8个供体。每一列误差线表示平均值 ± 标准差。(B)首先人T细胞被ImmunoCult™人CD3/CD28 T细胞激活剂活化3天,然后进 行电转。在电转48小时后,通过ArciTect™ T7核酸内切酶I试剂盒测定基因组编辑(切割)的效率。模拟电转:-RNP; RNP电转:+ RNP。(C - D)被ImmunoCult™人CD3/CD28 T细 胞激活剂活化3天的人T细胞经(C)模拟电转(没有RNP)和(D)RNP电转后TCRαβ和CD3的流式分析点图。(E)被ImmunoCult™人CD3/CD28 T细胞激活剂活化3天的人T细胞的 CD4和CD8流式分析点图。
激活后,用包含TRAC crRNA2的RNP复合物对T细胞进行电转。电转后细胞活率为52-82%(数据未显示),与激活条件无关。电转3天后(激活开
始后的第5或第6天)测定敲除效率,我们观察到敲除效率高达90%(TCRαβ细胞,图5A)。在激活3天后进行电转的敲除效率最高,尤其是在使用
ImmunoCult™人CD3/CD28 T细胞激活剂时(图5A)。此外,通过T7核酸内切酶I分析证实了来自所有4个供体经处理的样本中靶位点都有INDEL的形
成(图5B)。使用ImmunoCult™人CD3/CD28/CD2 T细胞激活剂培养2天后,供体之间的结果具有较大差异,其中一个样本没有反应。因此,我们使用
相同的条件测试了另外4个供体,并在这些样本中观察到较一致性的敲除结果。由于TCRαβ与CD3形成复合物,可通过在细胞表面上同时缺失CD3
的表达来确认基因敲除(图5A,C,D),而T细胞标记物CD4和CD8继续高表达(图5E)。
进行CRISPR-Cas9基因组编辑后,将细胞接种至含有IL-2的Immunocult™-XF T细胞扩增培养基中再培养10天,每2-3天进行换液。TCRαβ细胞在扩增
过程中一直存在(数据未显示)。相比于使用ImmunoCult™人CD3/CD28 T细胞激活剂处理的细胞,在电转前使用ImmunoCult™人CD3/CD28/CD2 T
细胞激活剂处理的细胞在基因编辑后显示出更高的扩增能力。电转后孵育7天,根据扩增倍数计算得出的细胞含量为4.1 x 107
至3.8 x 108
个细胞。
讨论
对原代人T细胞进行基因编辑时,细胞凋亡、细胞活率和基因编辑效率之间存在一定的平衡。我们发现电转前使用ImmunoCult™人CD3/CD28 T细
胞激活剂活化3天会得到最高且最一致的编辑效率。而用ImmunoCult™人CD3/CD28/CD2 T细胞激活剂刺激的细胞,在编辑后有更强的细胞扩增能
力。因此在选择操作流程时,应考虑下游应用所需要的细胞数量。如果下游应用需要大量编辑的细胞,那么可以在每次电转中按比例增加细胞或者
是增加更多的培养孔。通常,推荐使用两个孔作为阳性对照(例如ArciTect™人HPRT阳性对照试剂盒,产品号 #760134)和阴性对照(未电转)。应在
实验之前决定使用孔的数量以便计算需要的试剂总数。可能还需要考虑其他优化条件,例如细胞密度或Cas9:gRNA的比例,或者可能需要多个供
体样本(例如:患病和健康供体)。
本文所列举的基因组编辑策略适用于大多数基因敲除应用。特定应用的操作步骤调整(此处未详细介绍),可能包括:使用ArciTect™ Cas9-eGFP
核酸酶(产品号 #76005)进行阳性转染的可视化;单链核酸内切酶ArciTect™ Cas9(产品号 #76007)与两个邻近gRNA一起使用;或加入一个DNA
供体模板以进行同源性直接修复(HDR)介导的基因敲入。
参考文献
- Wang W et al. (2014) CCR gene disruption via lentiviral vectors expressing Cas9 and single guided RNA renders cells resistant to HIV-1 infection. PLoS One 9:e115987.
- Li C et al. (2015) Inhibition of HIV-1 infection of primary CD4+ T-cells by gene editing of CCR5 using adenovirus-delivered CRISPR/Cas9. J Gen Virol 96:2381–93.
- Mandal PK et al. (2014) Efficient ablation of genes in hematopoietic stem and effector cells using CRISPR/Cas9. J Gen Virol 163: 1817–26.
- Su S et al. (2016) CRISPR-Cas9 mediated efficient PD-1 disruption on human primary T cells from cancer patients. Sci Rep 6: 20070.
- Yin H et al. (2014) Non-viral vectors for gene-based therapy. Nat Rev Genet 15: 541–55.
- Hendel A et al. (2015) Chemically modified guide RNAs enhance CRISPR-Cas9 genome editing in human primary cells. Nat Biotechnol 33: 985–9.
- Schumann K et al. (2015) Generation of knock-in primary human T cells using Cas9 ribonucleoprotein. Proc Natl Acad Sci USA 112: 10437–42.
- Gomes-Silva D et al. (2017) CD7-edited T cells expressing a CD7-specific CAR for the therapy of T-cell malignancies. Blood 130: 285–96.
- Ren J et al. (2017) Multiplex genome editing to generate universal CAR T cells resistant to PD1 inhibition. Clin Cancer Res 23: 2255–66.
- Rupp LJ et al. (2017) CRISPR/Cas9-mediated PD-1 disruption enhances anti-tumor efficacy of human chimeric antigen receptor T cells. Sci Rep 7(1): 737.
- Kim S et al. (2018) CRISPR RNAs trigger innate immune responses in human cells. Genome Res 28: 1–7.
- Seki A & Rutz S (2018) Optimized RNP transfection for highly efficient CRISPR/Cas9-mediated gene knockout in primary T cells. J Exp Med 215(3): 985.
- Hou P et al. (2015) Genome editing of CXCR4 by CRISPR/Cas9 confers cells resistant to HIV-1 infection. Sci Rep 5: 15577.
- Hultquist JF (2016) A Cas9 Ribonucleoprotein Platform for Functional Genetic Studies of HIV-Host Interactions in Primary Human Cells. Cell Rep 17: 1438–52.
- Jung I-Y & Lee J (2018) Unleashing the therapeutic potential of CAR-T cell therapy using gene-editing technologies. Mol Cells 41(8): 717–23.
- Liu X et al. (2017) CRISPR-Cas9-mediated multiplex gene editing in CAR-T cells. Cell Res 27: 154–7.
- Ren J et al. (2017) A versatile system for rapid multiplex genome-edited CAR T cell generation. Oncotarget 8: 17002–11.
- Eyquem J et al. (2017) Targeting a CAR to the TRAC locus with CRISPR/Cas9 enhances tumour rejection. Nature 543(7643): 113–7.
- Haeussler M et al. (2016) Evaluation of off-target and on-target scoring algorithms and integration into the guide RNA selection tool CRISPOR. Genome Biol 17: 148.

 沪公网安备31010102008431号
沪公网安备31010102008431号