
 EasySep™小鼠TIL(CD45)正选试剂盒
EasySep™小鼠TIL(CD45)正选试剂盒




技术资料
-
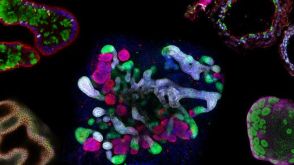 研究综述The Predictive Power of Organoid-Based New Approach Methodologies in Drug Discovery
研究综述The Predictive Power of Organoid-Based New Approach Methodologies in Drug Discovery细胞类型:
上皮细胞,多能干细胞,肠道细胞,肾细胞,胰腺细胞,PSC衍生上皮细胞,PSC衍生肝细胞,呼吸道细胞
发布日期: 10/31/2025 -
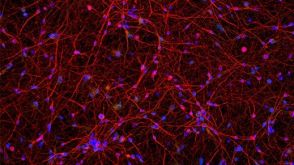 实验方案How to Culture Human Pluripotent Stem Cell (hPSC)-Derived Forebrain Neurons for MEA Analysis Using the Maestro MEA™ System
实验方案How to Culture Human Pluripotent Stem Cell (hPSC)-Derived Forebrain Neurons for MEA Analysis Using the Maestro MEA™ System研究方向:
干细胞生物学,疾病建模,神经科学,药物发现和毒性检测
发布日期: 11/05/2024 -
 31:49
线上讲座Using Human Pluripotent Stem Cell-Derived Neural Organoids for Disease Modeling发布日期: 08/01/2024
31:49
线上讲座Using Human Pluripotent Stem Cell-Derived Neural Organoids for Disease Modeling发布日期: 08/01/2024 -
 24:25
线上讲座ISSCR Standards in Action: Advancements in Cell Line Generation and Organoid Innovation发布日期: 08/01/2024
24:25
线上讲座ISSCR Standards in Action: Advancements in Cell Line Generation and Organoid Innovation发布日期: 08/01/2024









 沪公网安备31010102008431号
沪公网安备31010102008431号