
 EasySep™小鼠TIL(CD45)正选试剂盒
EasySep™小鼠TIL(CD45)正选试剂盒




技术资料
-

-
 专家访谈Scott Allen, PhD Seeking Metabolic Therapies for an Incurable Neurodegenerative Disease
专家访谈Scott Allen, PhD Seeking Metabolic Therapies for an Incurable Neurodegenerative Disease研究方向:
干细胞生物学,神经科学
发布日期: 08/29/2018 -
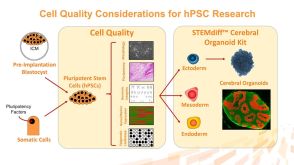
 技术手册Reversion and Maintenance of Human Naïve-Like Pluripotent Stem Cells with RSeT™ Medium发布日期: 08/01/2018
技术手册Reversion and Maintenance of Human Naïve-Like Pluripotent Stem Cells with RSeT™ Medium发布日期: 08/01/2018 -
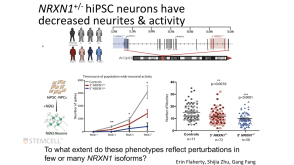
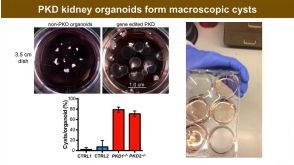
 技术公告Genome Editing with Direct Cas9 RNP Delivery Design Considerations
技术公告Genome Editing with Direct Cas9 RNP Delivery Design Considerations细胞类型:
T细胞,多能干细胞
发布日期: 06/01/2018



 沪公网安备31010102008431号
沪公网安备31010102008431号