Ye B-Q et al. (NOV 2010)
Journal of immunology (Baltimore,Md. : 1950) 185 10 6294--305
Slit2 regulates attractive eosinophil and repulsive neutrophil chemotaxis through differential srGAP1 expression during lung inflammation.
Directional migration of leukocytes is an essential step in leukocyte trafficking during inflammatory responses. However,the molecular mechanisms governing directional chemotaxis of leukocytes remain poorly understood. The Slit family of guidance cues has been implicated for inhibition of leuocyte migration. We report that Clara cells in the bronchial epithelium secreted Slit2,whereas eosinophils and neutrophils expressed its cell-surface receptor,Robo1. Compared to neutrophils,eosinophils exhibited a significantly lower level of Slit-Robo GTPase-activating protein 1 (srGAP1),leading to activation of Cdc42,recruitment of PI3K to Robo1,enhancment of eotaxin-induced eosinophil chemotaxis,and exaggeration of allergic airway inflammation. Notably,OVA sensitization elicited a Slit2 gradient at so-called bronchus-alveoli axis,with a higher level of Slit2 in the bronchial epithelium and a lower level in the alveolar tissue. Aerosol administration of rSlit2 accelerated eosinophil infiltration,whereas i.v. administered Slit2 reduced eosinophil deposition. In contrast,Slit2 inactivated Cdc42 and suppressed stromal cell-derived factor-1α-induced chemotaxis of neutrophils for inhibiting endotoxin-induced lung inflammation,which were reversed by blockade of srGAP1 binding to Robo1. These results indicate that the newly identified Slit2 gradient at the bronchus-alveoli axis induces attractive PI3K signaling in eosinophils and repulsive srGAP1 signaling in neutrophils through differential srGAP1 expression during lung inflammation.
View Publication
产品号#:
19256
19256RF
产品名:
Cowburn AS et al. (JUN 2011)
American journal of respiratory cell and molecular biology 44 6 879--87
Granulocyte/macrophage colony-stimulating factor causes a paradoxical increase in the BH3-only pro-apoptotic protein Bim in human neutrophils.
Neutrophil apoptosis is essential for the resolution of inflammation but is delayed by several inflammatory mediators. In such terminally differentiated cells it has been uncertain whether these agents can inhibit apoptosis through transcriptional regulation of anti-death (Bcl-X(L),Mcl-1,Bcl2A1) or BH3-only (Bim,Bid,Puma) Bcl2-family proteins. We report that granulocyte/macrophage colony-stimulating factor (GM-CSF) and tumor necrosis factor (TNF)-α prevent the normal time-dependent loss of Mcl-1 and Bcl2A1 in neutrophils,and we demonstrate that they cause an NF-κB-dependent increase in Bcl-X(L) transcription/translation. We show that GM-CSF and TNF-α increase and/or maintain mRNA levels for the pro-apoptotic BH3-only protein Bid and that GM-CSF has a similar NF-κB-dependent effect on Bim transcription and BimEL expression. The in-vivo relevance of these findings was indicated by demonstrating that GM-CSF is the dominant neutrophil survival factor in lung lavage from patients with ventilator-associated pneumonia,confirming an increase in lung neutrophil Bim mRNA. Finally GM-CSF caused mitochondrial location of Bim and a switch in phenotype to a cell that displays accelerated caspase-9-dependent apoptosis. This study demonstrates the capacity of neutrophil survival agents to induce a paradoxical increase in the pro-apoptotic proteins Bid and Bim and suggests that this may function to facilitate rapid apoptosis at the termination of the inflammatory cycle.
View Publication
产品号#:
19257
19257RF
21000
20119
20155
产品名:
RoboSep™- S
RoboSep™ 吸头组件抛光剂
RoboSep™分选管套装(9个塑料管)
Velu CS et al. (MAY 2009)
Blood 113 19 4720--8
Gfi1 regulates miR-21 and miR-196b to control myelopoiesis.
The zinc finger protein growth factor independent-1 (Gfi1) is a transcriptional repressor that is critically required for normal granulocytic differentiation. GFI1 loss-of-function mutations are found in some patients with severe congenital neutropenia (SCN). The SCN-associated GFI1-mutant proteins act as dominant negatives to block granulopoiesis through selective deregulation of a subset of GFI1 target genes. Here we show that Gfi1 is a master regulator of microRNAs,and that deregulated expression of these microRNAs recapitulates a Gfi1 loss-of-function block to granulocyte colony-stimulating factor (G-CSF)-stimulated granulopoiesis. Specifically,bone marrow cells from a GFI1-mutant SCN patient and Gfi1(-/-) mice display deregulated expression of miR-21 and miR-196B expression. Flow cytometric analysis and colony assays reveal that the overexpression or depletion of either miR induces changes in myeloid development. However,coexpression of miR-21 and miR-196b (as seen in Gfi1(-/-) mice and a GFI1N382S SCN patient) completely blocks G-CSF-induced granulopoiesis. Thus,our results not only identify microRNAs whose regulation is required during myelopoiesis,but also provide an example of synergy in microRNA biologic activity and illustrate potential mechanisms underlying SCN disease pathogenesis.
View Publication
产品号#:
03534
09600
09650
产品名:
MethoCult™ GF M3534
StemSpan™ SFEM
StemSpan™ SFEM
Eash KJ et al. (MAY 2009)
Blood 113 19 4711--9
CXCR4 is a key regulator of neutrophil release from the bone marrow under basal and stress granulopoiesis conditions.
The number of neutrophils in the blood is tightly regulated to ensure adequate protection against microbial pathogens while minimizing damage to host tissue. Neutrophil homeostasis in the blood is achieved through a balance of neutrophil production,release from the bone marrow,and clearance from the circulation. Accumulating evidence suggests that signaling by CXCL12,through its major receptor CXCR4,plays a key role in maintaining neutrophil homeostasis. Herein,we generated mice with a myeloid lineage-restricted deletion of CXCR4 to define the mechanisms by which CXCR4 signals regulate this process. We show that CXCR4 negatively regulates neutrophil release from the bone marrow in a cell-autonomous fashion. However,CXCR4 is dispensable for neutrophil clearance from the circulation. Neutrophil mobilization responses to granulocyte colony-stimulating factor (G-CSF),CXCL2,or Listeria monocytogenes infection are absent or impaired,suggesting that disruption of CXCR4 signaling may be a common step mediating neutrophil release. Collectively,these data suggest that CXCR4 signaling maintains neutrophil homeostasis in the blood under both basal and stress granulopoiesis conditions primarily by regulating neutrophil release from the bone marrow.
View Publication
产品号#:
03434
03444
产品名:
MethoCult™ GF M3434
MethoCult™ GF M3434
Fan Y et al. (JAN 2018)
The Biochemical journal 475 1 23--44
Interrogating Parkinson's disease LRRK2 kinase pathway activity by assessing Rab10 phosphorylation in human neutrophils.
There is compelling evidence for the role of the leucine-rich repeat kinase 2 (LRRK2) and in particular its kinase function in Parkinson's disease. Orally bioavailable,brain penetrant and potent LRRK2 kinase inhibitors are in the later stages of clinical development. Here,we describe a facile and robust assay to quantify LRRK2 kinase pathway activity by measuring LRRK2-mediated phosphorylation of Rab10 in human peripheral blood neutrophils. We use the selective MJFF-pRab10 monoclonal antibody recognising the Rab10 Thr73 phospho-epitope that is phosphorylated by LRRK2. We highlight the feasibility and practicability of using our assay in the clinical setting by studying a few patients with G2019S LRRK2 associated and sporadic Parkinson's as well as healthy controls. We suggest that peripheral blood neutrophils are a valuable resource for LRRK2 research and should be considered for inclusion in Parkinson's bio-repository collections as they are abundant,homogenous and express relatively high levels of LRRK2 as well as Rab10. In contrast,the widely used peripheral blood mononuclear cells are heterogeneous and only a minority of cells (monocytes and contaminating neutrophils) express LRRK2. While our LRRK2 kinase pathway assay could assist in patient stratification based on LRRK2 kinase activity,we envision that it may find greater utility in pharmacodynamic and target engagement studies in future LRRK2 inhibitor trials.
View Publication
Verma AH et al. (APR 2016)
Mucosal immunology April 1--11
Eosinophils subvert host resistance to an intracellular pathogen by instigating non-protective IL-4 in CCR2(-/-) mice.
Eosinophils contribute to type II immune responses in helminth infections and allergic diseases; however,their influence on intracellular pathogens is less clear. We previously reported that CCR2(-/-) mice exposed to the intracellular fungal pathogen Histoplasma capsulatum exhibit dampened immunity caused by an early exaggerated interleukin (IL)-4 response. We sought to identify the cellular source promulgating IL-4 in infected mutant animals. Eosinophils were the principal instigators of non-protective IL-4 and depleting this granulocyte population improved fungal clearance in CCR2(-/-) animals. The deleterious impact of eosinophilia on mycosis was also recapitulated in transgenic animals overexpressing eosinophils. Mechanistic examination of IL-4 induction revealed that phagocytosis of H. capsulatum via the pattern recognition receptor complement receptor (CR) 3 triggered the heightened IL-4 response in murine eosinophils. This phenomenon was conserved in human eosinophils; exposure of cells to the fungal pathogen elicited a robust IL-4 response. Thus,our findings elucidate a detrimental attribute of eosinophil biology in fungal infections that could potentially trigger a collapse in host defenses by instigating type II immunity.Mucosal Immunology advance online publication,6 April 2016; doi:10.1038/mi.2016.26.
View Publication
产品号#:
19256
19256RF
产品名:
Brode S et al. (DEC 2010)
Thorax 65 12 1116--7
Interleukin-5 inhibits glucocorticoid-mediated apoptosis in human eosinophils.
Mentlik AN et al. (JUL 2010)
Molecular biology of the cell 21 13 2241--56
Rapid lytic granule convergence to the MTOC in natural killer cells is dependent on dynein but not cytolytic commitment.
Natural killer cells are lymphocytes specialized to participate in host defense through their innate ability to mediate cytotoxicity by secreting the contents of preformed secretory lysosomes (lytic granules) directly onto a target cell. This form of directed secretion requires the formation of an immunological synapse and occurs stepwise with actin reorganization preceding microtubule-organizing center (MTOC) polarization to the synapse. Because MTOC polarization to the synapse is required for polarization of lytic granules,we attempted to define their interrelationship. We found that compared with the time required for MTOC polarization,lytic granules converged to the MTOC rapidly. The MTOC-directed movement of lytic granules was independent of actin and microtubule reorganization,dependent on dynein motor function,occurred before MTOC polarization,and did not require a commitment to cytotoxicity. This defines a novel paradigm for rapid MTOC-directed transport as a prerequisite for directed secretion,one that may prepare,but not commit cells for precision secretory function.
View Publication
产品号#:
15025
15065
产品名:
RosetteSep™人NK细胞富集抗体混合物
RosetteSep™人NK细胞富集抗体混合物
Hornick EE et al. (FEB 2018)
Journal of immunology (Baltimore,Md. : 1950) 200 3 1188--1197
Nlrp12 Mediates Adverse Neutrophil Recruitment during Influenza Virus Infection.
Exaggerated inflammatory responses during influenza A virus (IAV) infection are typically associated with severe disease. Neutrophils are among the immune cells that can drive this excessive and detrimental inflammation. In moderation,however,neutrophils are necessary for optimal viral control. In this study,we explore the role of the nucleotide-binding domain leucine-rich repeat containing receptor family member Nlrp12 in modulating neutrophilic responses during lethal IAV infection. Nlrp12-/- mice are protected from lethality during IAV infection and show decreased vascular permeability,fewer pulmonary neutrophils,and a reduction in levels of neutrophil chemoattractant CXCL1 in their lungs compared with wild-type mice. Nlrp12-/- neutrophils and dendritic cells within the IAV-infected lungs produce less CXCL1 than their wild-type counterparts. Decreased CXCL1 production by Nlrp12-/- dendritic cells was not due to a difference in CXCL1 protein stability,but instead to a decrease in Cxcl1 mRNA stability. Together,these data demonstrate a previously unappreciated role for Nlrp12 in exacerbating the pathogenesis of IAV infection through the regulation of CXCL1-mediated neutrophilic responses.
View Publication
产品号#:
19762
19762RF
产品名:
EasySep™小鼠中性粒细胞富集试剂盒
RoboSep™ 小鼠中性粒细胞富集试剂盒含滤芯吸头
C. R. Seehus et al. (DEC 2017)
Nature communications 8 1 1900
Alternative activation generates IL-10 producing type 2 innate lymphoid cells.
Type 2 innate lymphoid cells (ILC2) share cytokine and transcription factor expression with CD4+ Th2 cells,but functional diversity of the ILC2 lineage has yet to be fully explored. Here,we show induction of a molecularly distinct subset of activated lung ILC2,termed ILC210. These cells produce IL-10 and downregulate some pro-inflammatory genes. Signals that generate ILC210 are distinct from those that induce IL-13 production,and gene expression data indicate that an alternative activation pathway leads to the generation of ILC210. In vivo,IL-2 enhances ILC210 generation and is associated with decreased eosinophil recruitment to the lung. Unlike most activated ILC2,the ILC210 population contracts after cessation of stimulation in vivo,with maintenance of a subset that can be recalled by restimulation,analogous to T-cell effector cell and memory cell generation. These data demonstrate the generation of a previously unappreciated IL-10 producing ILC2 effector cell population.
View Publication
产品号#:
19860
19860RF
85415
85420
85450
85460
86415
86420
86450
86460
产品名:
EasySep™小鼠Streptavidin RapidSpheres™分选试剂盒
RoboSep™ 小鼠Streptavidin RapidSpheres™分选试剂盒
SepMate™-15 (IVD)
SepMate™-15 (IVD)
SepMate™-50 (IVD)
SepMate™-50 (IVD)
SepMate™-15 (RUO)
SepMate™-15 (RUO)
SepMate™-50 (RUO)
SepMate™-50 (RUO)
Ode Y et al. (JAN 2018)
Journal of leukocyte biology
CIRP increases ICAM-1+ phenotype of neutrophils exhibiting elevated iNOS and NETs in sepsis.
Sepsis represents uncontrolled inflammation due to an infection. Cold-inducible RNA-binding protein (CIRP) is a stress-induced damage-associated molecular pattern (DAMP). A subset of neutrophils expressing ICAM-1+ neutrophils was previously shown to produce high levels of reactive oxygen species. The role of CIRP for the development and function of ICAM-1+ neutrophils during sepsis is unknown. We hypothesize that CIRP induces ICAM-1 expression in neutrophils causing injury to the lungs during sepsis. Using a mouse model of cecal ligation and puncture (CLP)-induced sepsis,we found increased expression of CIRP and higher frequencies and numbers of ICAM-1+ neutrophils in the lungs. Conversely,the CIRP-/- mice showed significant inhibition in the frequencies and numbers of ICAM-1+ neutrophils in the lungs compared to wild-type (WT) mice in sepsis. In vitro treatment of bone marrow-derived neutrophils (BMDN) with recombinant murine CIRP (rmCIRP) significantly increased ICAM-1+ phenotype in a time- and dose-dependent manner. The effect of rmCIRP on increasing frequencies of ICAM-1+ neutrophils was significantly attenuated in BMDN treated with anti-TLR4 Ab or NF-κB inhibitor compared,respectively,with BMDN treated with isotype IgG or DMSO. The frequencies of iNOS producing and neutrophil extracellular traps (NETs) forming phenotypes in rmCIRP-treated ICAM-1+ BMDN were significantly higher than those in ICAM-1- BMDN. Following sepsis the ICAM-1+ neutrophils in the lungs showed significantly higher levels of iNOS and NETs compared to ICAM-1- neutrophils. We further revealed that ICAM-1 and NETs were co-localized in the neutrophils treated with rmCIRP. CIRP-/- mice showed significant improvement in their survival outcome (78% survival) over that of WT mice (48% survival) in sepsis. Thus,CIRP could be a novel therapeutic target for regulating iNOS producing and NETs forming ICAM-1+ neutrophils in the lungs during sepsis.
View Publication

 EasySep™小鼠TIL(CD45)正选试剂盒
EasySep™小鼠TIL(CD45)正选试剂盒




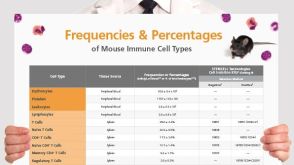 挂图Frequencies and Percentages of Mouse Immune Cell Types List of the frequencies of over 25 immune cell types in C57BL/6 mice
挂图Frequencies and Percentages of Mouse Immune Cell Types List of the frequencies of over 25 immune cell types in C57BL/6 mice
 沪公网安备31010102008431号
沪公网安备31010102008431号