
 EasySep™小鼠TIL(CD45)正选试剂盒
EasySep™小鼠TIL(CD45)正选试剂盒




技术资料
-
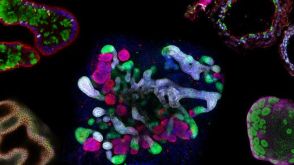 研究综述The Predictive Power of Organoid-Based New Approach Methodologies in Drug Discovery
研究综述The Predictive Power of Organoid-Based New Approach Methodologies in Drug Discovery细胞类型:
上皮细胞,多能干细胞,肠道细胞,胰腺细胞,肾脏细胞,PSC衍生上皮细胞,PSC衍生肝细胞,呼吸道细胞
-

-

-
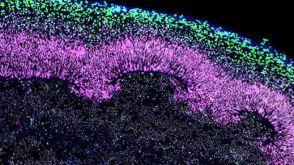
 科学海报A Reliable, Efficient, and Matrix-Free Method to Generate Midbrain Organoids from Human Pluripotent Stem Cells
科学海报A Reliable, Efficient, and Matrix-Free Method to Generate Midbrain Organoids from Human Pluripotent Stem CellsConference:
CSHL 3D Brain 2022
-
-
 科学海报An In Vitro Assay to Determine the Neurotoxic Effects of Pharmacological Compounds
科学海报An In Vitro Assay to Determine the Neurotoxic Effects of Pharmacological CompoundsConference:
SfN 2022







 沪公网安备31010102008431号
沪公网安备31010102008431号