Human milk oligosaccharides differentially support gut barrier integrity and enhance Th1 and Th17 cell effector responses
Human milk oligosaccharides (HMOs) can modulate the intestinal barrier and regulate immune cells to favor the maturation of the infant intestinal tract and immune system,but the precise functions of individual HMOs are unclear. To determine the structure-dependent effects of individual HMOs (representing different structural classes) on the intestinal epithelium as well as innate and adaptive immune cells,we assessed fucosylated (2′FL and 3FL),sialylated (3′SL and 6′SL) and neutral non-fucosylated (LNT and LNT2) HMOs for their ability to support intestinal barrier integrity,to stimulate the secretion of chemokines from intestinal epithelial cells,and to modulate cytokine release from LPS-activated dendritic cells (DCs),M1 macrophages (MØs),and co-cultures with naïve CD4+ T cells. The fucosylated and neutral non-fucosylated HMOs increased barrier integrity and protected the barrier following an inflammatory insult but exerted minimal immunomodulatory activity. The sialylated HMOs enhanced the secretion of CXCL10,CCL20 and CXCL8 from intestinal epithelial cells,promoted the secretion of several cytokines (including IL-10,IL-12p70 and IL-23) from LPS-activated DCs and M1 MØs,and increased the secretion of IFN-γ and IL-17A from CD4+ T cells primed by LPS-activated DCs and MØs while reducing the secretion of IL-13. Thus,3′SL and 6′SL supported Th1 and Th17 responses while reducing Th2 responses. Collectively,our data show that HMOs exert structure-dependent effects on the intestinal epithelium and possess immunomodulatory properties that confer benefits to infants and possibly also later in life.
View Publication
产品号#:
18000
19359
100-0697
19359RF
产品名:
EasySep™磁极
EasySep™人单核细胞分选试剂盒
EasySep™人单核细胞分选试剂盒
RoboSep™ 人单核细胞分选试剂盒
(Feb 2024)
Frontiers in Oncology 14 8
A FACS-based novel isolation technique identifies heterogeneous CTCs in oral squamous cell carcinoma
PurposeIsolating circulating tumour cells (CTCs) from the blood is challenging due to their low abundance and heterogeneity. Limitations of conventional CTC detection methods highlight the need for improved strategies to detect and isolate CTCs. Currently,the Food and Drug Administration (FDA)-approved CellSearch™ and other RUO techniques are not available in India. Therefore,we wanted to develop a flexible CTC detection/isolation technique that addresses the limitation(s) of currently available techniques and is suitable for various downstream applications.MethodsWe developed a novel,efficient,user-friendly CTC isolation strategy combining density gradient centrifugation and immuno-magnetic hematogenous cell depletion with fluorescence-activated cell sorting (FACS)-based positive selection using multiple CTC-specific cell-surface markers. For FACS,a stringent gating strategy was optimised to exclude debris and doublets by side scatter/forward scatter (SSC/FSC) discriminator,remove dead cells by 4′,6-diamidino-2-phenylindole (DAPI) staining,and eliminate non-specific fluorescence using a “dump” channel. APC-labelled anti-CD45mAB was used to gate remaining hematogenous cells,while multiple epithelial markers (EpCAM,EGFR,and Pan-Cytokeratin) and an epithelial–mesenchymal transition (EMT) marker (Vimentin) labelled with fluorescein isothiocyanate (FITC) were used to sort cancer cells. The technique was initially developed by spiking Cal 27 cancer cells into the blood of healthy donors and then validated in 95 biopsy-proven oral squamous cell carcinoma (OSCC) patients. CTCs isolated from patients were reconfirmed by Giemsa staining,immuno-staining,and whole transcriptome amplification (WTA),followed by qRT-PCR. In vitro culture and RNA sequencing (RNA-Seq) were also performed to confirm their suitability for various downstream applications.ResultsThe mean detection efficiency for the Cal 27 tongue cancer cells spiked in the whole blood of healthy donors was 32.82% ± 12.71%. While ~75% of our patients (71/95) had detectable CTCs,the CTC positivity was independent of the TNM staging. The isolated potential cancer cells from OSCC patients were heterogeneous in size. They expressed different CTC-specific markers in various combinations as identified by qRT-PCR after WTA in different patients. Isolated CTCs were also found to be suitable for downstream applications like short-term CTC culture and RNA-Seq.ConclusionWe developed a sensitive,specific,flexible,and affordable CTC detection/isolation technique,which is scalable to larger patient cohorts,provides a snapshot of CTC heterogeneity,isolates live CTCs ready for downstream molecular analysis,and,most importantly,is suitable for developing countries.
View Publication
产品号#:
17898
17898RF
产品名:
EasySep™人CD45去除试剂盒II
RoboSep™ 人CD45去除试剂盒II
(Mar 2024)
The European Respiratory Journal 63 3
Extensive acute and sustained changes to neutrophil proteomes post-SARS-CoV-2 infection
Graphical abstract Summary of the study. Peripheral blood neutrophils from >200 hospitalised patients across three patient groups (coronavirus disease 2019 (COVID-19),non-COVID-19 lower respiratory tract infection (LRTI) and matched controls) were comprehensively profiled using mass spectrometry,revealing novel proteomic changes in acute and convalescent COVID-19. DIA: data-independent acquisition; TLR: Toll-like receptor; ARG: arginase; TGF: transforming growth factor; IFN: interferon. BackgroundNeutrophils are important in the pathophysiology of coronavirus disease 2019 (COVID-19),but the molecular changes contributing to altered neutrophil phenotypes following severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2) infection are not fully understood. We used quantitative mass spectrometry-based proteomics to explore neutrophil phenotypes immediately following acute SARS-CoV-2 infection and during recovery.MethodsProspective observational study of hospitalised patients with PCR-confirmed SARS-CoV-2 infection (May to December 2020). Patients were enrolled within 96 h of admission,with longitudinal sampling up to 29 days. Control groups comprised non-COVID-19 acute lower respiratory tract infection (LRTI) and age-matched noninfected controls. Neutrophils were isolated from peripheral blood and analysed using mass spectrometry. COVID-19 severity and recovery were defined using the World Health Organization ordinal scale.ResultsNeutrophil proteomes from 84 COVID-19 patients were compared to those from 91 LRTI and 42 control participants. 5800 neutrophil proteins were identified,with >1700 proteins significantly changed in neutrophils from COVID-19 patients compared to noninfected controls. Neutrophils from COVID-19 patients initially all demonstrated a strong interferon signature,but this signature rapidly declined in patients with severe disease. Severe disease was associated with increased abundance of proteins involved in metabolism,immunosuppression and pattern recognition,while delayed recovery from COVID-19 was associated with decreased granule components and reduced abundance of metabolic proteins,chemokine and leukotriene receptors,integrins and inhibitory receptors.ConclusionsSARS-CoV-2 infection results in the sustained presence of circulating neutrophils with distinct proteomes suggesting altered metabolic and immunosuppressive profiles and altered capacities to respond to migratory signals and cues from other immune cells,pathogens or cytokines. Tweetable abstractHigh-resolution mass spectrometry analysis of peripheral blood neutrophils from >200 individuals provides novel insights into neutrophil phenotypes during acute COVID-19 and reveals that altered neutrophils persist in convalescent COVID-19 patients https://bit.ly/3QSSq9W
View Publication
产品号#:
19666
100-0404
产品名:
EasySep™ Direct人中性粒细胞分选试剂盒
RoboSep™ 人中性粒细胞分选试剂盒
(Feb 2024)
iScience 27 3
A1-reprogrammed mesenchymal stromal cells prime potent antitumoral responses
SummaryMesenchymal stromal cells (MSCs) have been modified via genetic or pharmacological engineering into potent antigen-presenting cells-like capable of priming responding CD8 T cells. In this study,our screening of a variant library of Accum molecule revealed a molecule (A1) capable of eliciting antigen cross-presentation properties in MSCs. A1-reprogrammed MSCs (ARM) exhibited improved soluble antigen uptake and processing. Our comprehensive analysis,encompassing cross-presentation assays and molecular profiling,among other cellular investigations,elucidated A1’s impact on endosomal escape,reactive oxygen species production,and cytokine secretion. By evaluating ARM-based cellular vaccine in mouse models of lymphoma and melanoma,we observe significant therapeutic potency,particularly in allogeneic setting and in combination with anti-PD-1 immune checkpoint inhibitor. Overall,this study introduces a strong target for developing an antigen-adaptable vaccination platform,capable of synergizing with immune checkpoint blockers to trigger tumor regression,supporting further investigation of ARMs as an effective and versatile anti-cancer vaccine. Graphical abstract Highlights•Treatment with A1/antigen mix reprograms MSCs into antigen-presenting cells•The antigen cross-presenting ability of ARM cells require ROS and UPR•ARMs synergize with immune-checkpoint inhibitors in priming potent antitumoral activity Classification Description: Immunology; Pharmaceutical engineering; Cancer
View Publication
产品号#:
18953
18953RF
产品名:
EasySep™小鼠CD8a正选试剂盒II
RoboSep™ 小鼠CD8a正选试剂盒II
(Feb 2024)
Nature Communications 15
Comprehensive characterization of IFNγ signaling in acute myeloid leukemia reveals prognostic and therapeutic strategies
Interferon gamma (IFNγ) is a critical cytokine known for its diverse roles in immune regulation,inflammation,and tumor surveillance. However,while IFNγ levels were elevated in sera of most newly diagnosed acute myeloid leukemia (AML) patients,its complex interplay in AML remains insufficiently understood. We aim to characterize these complex interactions through comprehensive bulk and single-cell approaches in bone marrow of newly diagnosed AML patients. We identify monocytic AML as having a unique microenvironment characterized by IFNγ producing T and NK cells,high IFNγ signaling,and immunosuppressive features. IFNγ signaling score strongly correlates with venetoclax resistance in primary AML patient cells. Additionally,IFNγ treatment of primary AML patient cells increased venetoclax resistance. Lastly,a parsimonious 47-gene IFNγ score demonstrates robust prognostic value. In summary,our findings suggest that inhibiting IFNγ is a potential treatment strategy to overcoming venetoclax resistance and immune evasion in AML patients. IFNγ signaling is important in the pathogenesis and immune response,emphasizing the need for investigation of its role. Here,the authors show that IFNγ plays a key role in shaping immune microenvironment in AML and developing resistance,providing insights for potential therapeutic strategies.
View Publication
产品号#:
17858
17856
17858RF
100-0694
17856RF
100-1569
产品名:
EasySep™人CD14正选试剂盒II
EasySep™人CD34正选试剂盒 II
RoboSep™ 人CD14正选试剂盒II
EasySep™人CD14正选试剂盒II
EasySep™人CD34正选试剂盒 II
EasySep™人CD34正选试剂盒 II
(Feb 2024)
Pathogens and Immunity 8 2
HIV Productively Infects Highly Differentiated and Exhausted CD4+ T Cells During AIDS
Background:Throughout HIV infection,productively infected cells generate billions of viral particles and are thus responsible for body-wide HIV dissemination,but their phenotype during AIDS is unknown. As AIDS is associated with immunological changes,analyzing the phenotype of productively infected cells can help understand HIV production during this terminal stage.Methods:Blood samples from 15 untreated viremic participants (recent infection,n=5; long-term infection,n=5; active opportunistic AIDS-defining disease,n=5) and 5 participants virologically controlled on antiretroviral therapy (ART) enrolled in the Analysis of the Persistence,Reservoir and HIV Latency (APRIL) study (NCT05752318) were analyzed. Cells expressing the capsid protein p24 (p24+ cells) after 18 hours of resting or 24 hours of stimulation (HIV-Flow) revealed productively infected cells from viremic participants or translation-competent reservoir cells from treated participants,respectively.Results:The frequency of productively infected cells tended to be higher during AIDS in comparison with recent and long-term infections (median,340,72,and 32/million CD4+ T cells,respectively) and correlated with the plasma viral load at all stages of infection. Altogether,these cells were more frequently CD4low,HLA-ABClow,CD45RA-,Ki67+,PD-1+,with a non-negligible contribution from pTfh (CXCR5+PD-1+) cells,and were not significantly enriched in HIV coreceptors CCR5 nor CXCR4 expression. The comparison markers expression between stages showed that productively infected cells during AIDS were enriched in memory and exhausted cells. In contrast,the frequencies of infected pTfh were lower during AIDS compared to non-AIDS stages. A UMAP analysis revealed that total CD4+ T cells were grouped in 7 clusters and that productive p24+ cells were skewed to given clusters throughout the course of infection. Overall,the preferential targets of HIV during the latest stages seemed to be more frequently highly differentiated (memory,TTD-like) and exhausted cells and less frequently pTfh-like cells. In contrast,translation-competent reservoir cells were less frequent (5/million CD4+ T cells) and expressed more frequently HLA-ABC and less frequently PD-1.Conclusions:In long-term infection and AIDS,productively infected cells were differentiated and exhausted. This could indicate that cells with these given features are responsible for HIV production and dissemination in an immune dysfunction environment occurring during the last stages of infection.
View Publication
产品号#:
19052
19052RF
产品名:
EasySep™人CD4+ T细胞富集试剂盒
RoboSep™ 人CD4+ T细胞富集试剂盒含滤芯吸头
(Feb 2024)
Cell Reports Medicine 5 2
Preclinical characterization and phase 1 results of ADG106 in patients with advanced solid tumors and non-Hodgkin’s lymphoma
SummaryADG106,a ligand-blocking agonistic antibody targeting CD137 (4-1BB),exhibits promising results in preclinical studies,demonstrating tumor suppression in various animal models and showing a balanced profile between safety and efficacy. This phase 1 study enrolls 62 patients with advanced malignancies,revealing favorable tolerability up to the 5.0 mg/kg dose level. Dose-limiting toxicity occurs in only one patient (6.3%) at 10.0 mg/kg,resulting in grade 4 neutropenia. The most frequent treatment-related adverse events include leukopenia (22.6%),neutropenia (22.6%),elevated alanine aminotransferase (22.6%),rash (21.0%),itching (17.7%),and elevated aspartate aminotransferase (17.7%). The overall disease control rates are 47.1% for advanced solid tumors and 54.5% for non-Hodgkin’s lymphoma. Circulating biomarkers suggest target engagement by ADG106 and immune modulation of circulating T,B,and natural killer cells and cytokines interferon γ and interleukin-6,which may affect the probability of clinical efficacy. ADG106 has a manageable safety profile and preliminary anti-tumor efficacy in patients with advanced cancers (this study was registered at ClinicalTrials.gov: NCT03802955). Graphical abstract Highlights•ADG106 is a ligand-blocking agonistic antibody targeting CD137•ADG106 enhances cytotoxic T cell activity within the tumor environment•ADG106 shows manageable safety and preliminary anti-tumor efficacy in this phase 1 study Ma et al. demonstrate the safety,efficacy,and survival benefits of ADG106,a fully human agonistic monoclonal IgG4 antibody targeting a unique and crossreactive epitope of CD137,in patients with advanced solid tumors and non-Hodgkin’s lymphoma. They show that ADG106 exhibits a favorable safety profile and encourages anti-tumor activity.
View Publication
产品号#:
17953
17953RF
100-0710
产品名:
EasySep™人CD8+ T细胞分选试剂盒
RoboSep™ 人CD8+ T细胞分选试剂盒
EasySep™人CD8+ T细胞分选试剂盒
(Feb 2024)
Nature Communications 15
Serum amyloid A promotes glycolysis of neutrophils during PD-1 blockade resistance in hepatocellular carcinoma
The response to programmed death-1 (PD-1) blockade varies in hepatocellular carcinoma (HCC). We utilize a panel of 16 serum factors to show that a circulating level of serum amyloid A (SAA) > 20.0 mg/L has the highest accuracy in predicting anti-PD-1 resistance in HCC. Further experiments show a correlation between peritumoral SAA expression and circulating SAA levels in patients with progressive disease after PD-1 inhibition. In vitro experiments demonstrate that SAA induces neutrophils to express PD-L1 through glycolytic activation via an LDHA/STAT3 pathway and to release oncostatin M,thereby attenuating cytotoxic T cell function. In vivo,genetic or pharmacological inhibition of STAT3 or SAA eliminates neutrophil-mediated immunosuppression and enhances antitumor efficacy of anti-PD-1 treatment. This study indicates that SAA may be a critical inflammatory cytokine implicated in anti-PD-1 resistance in HCC. Targeting SAA-induced PD-L1+ neutrophils through STAT3 or SAA inhibition may present a potential approach for overcoming anti-PD1 resistance. The reasons for why hepatocellular carcinoma (HCC) is unresponsive to anti-PD-1 inhibition in some patients is not fully understood. Here the authors use human samples and mice tumour models to implicate serum amyloid A and STAT3 signalling involvement in the resistance to anti-PD1 immunotherapy in HCC.
View Publication
产品号#:
19666
17853
100-0404
17853RF
100-0699
产品名:
EasySep™ Direct人中性粒细胞分选试剂盒
EasySep™人CD8正选试剂盒 II
RoboSep™ 人中性粒细胞分选试剂盒
RoboSep™ 人CD8正选试剂盒 II
EasySep™人CD8阳性选择试剂盒II
(Feb 2024)
Nature Communications 15
Structure-guided engineering of immunotherapies targeting TRBC1 and TRBC2 in T cell malignancies
Peripheral T cell lymphomas are typically aggressive with a poor prognosis. Unlike other hematologic malignancies,the lack of target antigens to discriminate healthy from malignant cells limits the efficacy of immunotherapeutic approaches. The T cell receptor expresses one of two highly homologous chains [T cell receptor β-chain constant (TRBC) domains 1 and 2] in a mutually exclusive manner,making it a promising target. Here we demonstrate specificity redirection by rational design using structure-guided computational biology to generate a TRBC2-specific antibody (KFN),complementing the antibody previously described by our laboratory with unique TRBC1 specificity (Jovi-1) in targeting broader spectrum of T cell malignancies clonally expressing either of the two chains. This permits generation of paired reagents (chimeric antigen receptor-T cells) specific for TRBC1 and TRBC2,with preclinical evidence to support their efficacy in T cell malignancies. The T cell receptor β-chain is expressed in two isoforms,TRBC1 and TRBC2,with clonally expanded mature T cell lymphomas expressing one of them exclusively,while healthy T cells randomly express either TRBC1 or TRBC2. Here authors show structure-based design of a TRBC2-specific antibody,and depletion of malignant T cells carrying TRBC1 or TRBC2 with CAR-T cells against the cognate receptor chain in murine models.
View Publication

 EasySep™小鼠TIL(CD45)正选试剂盒
EasySep™小鼠TIL(CD45)正选试剂盒




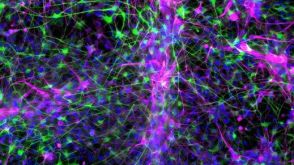 实验方案How to Co-Culture Astrocytes and NGN2 mRNA-Driven Induced Forebrain Neurons Derived from Human Pluripotent Stem Cells
实验方案How to Co-Culture Astrocytes and NGN2 mRNA-Driven Induced Forebrain Neurons Derived from Human Pluripotent Stem Cells

 沪公网安备31010102008431号
沪公网安备31010102008431号